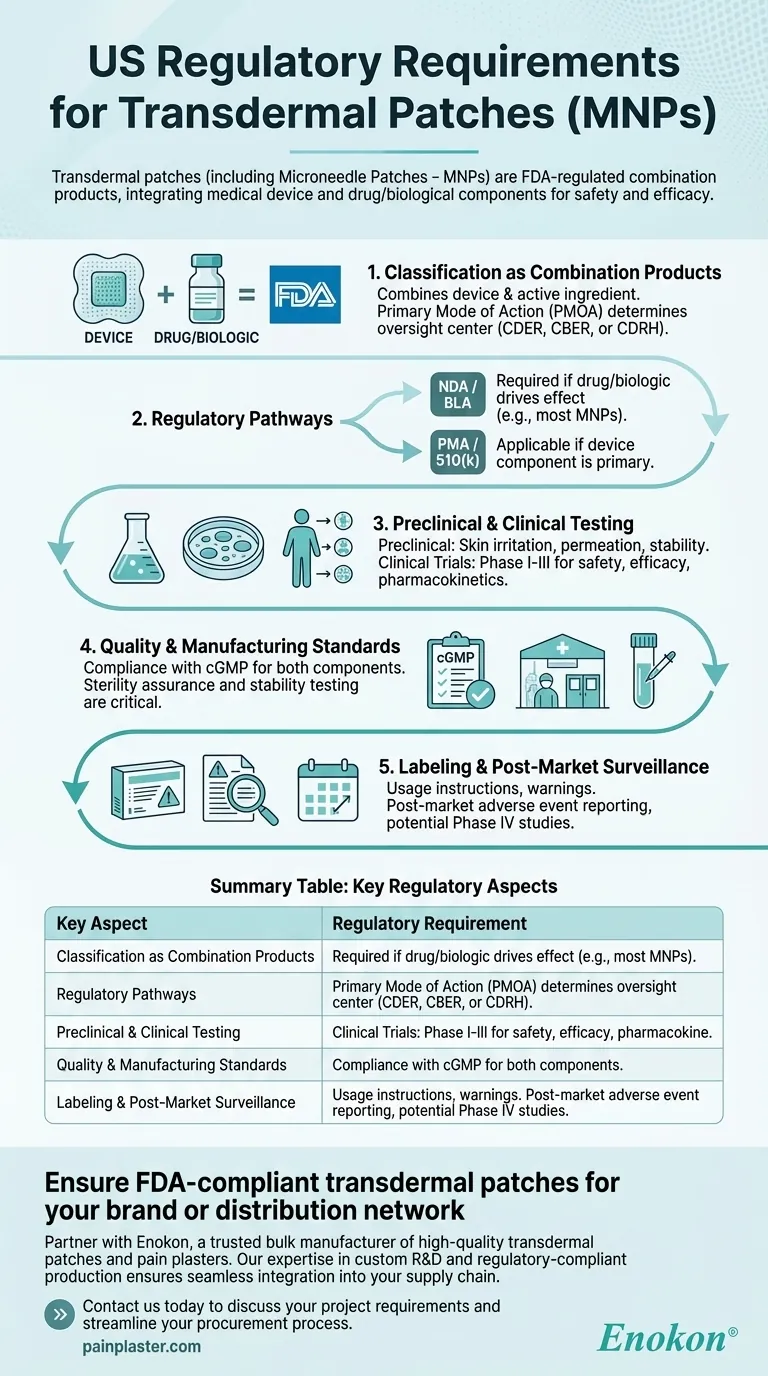
Negli Stati Uniti i cerotti transdermici, compresi i cerotti con microaghi (MNP), sono regolamentati dalla FDA come prodotti combinati e richiedono processi di approvazione rigorosi per garantire sicurezza ed efficacia. Questi prodotti integrano componenti di dispositivi medici e di farmaci/biologici, richiedendo la conformità a percorsi normativi specifici a seconda della loro modalità d'azione primaria. Il processo di approvazione prevede test preclinici e clinici, controlli di qualità e il rispetto degli standard di etichettatura e produzione.
Punti chiave spiegati:
-
Classificazione come prodotti di combinazione
- La FDA classifica cerotti transdermici come prodotti combinati in quanto combinano un dispositivo medico (ad esempio, il supporto del cerotto, i microneedles) con un farmaco o un principio attivo biologico.
- La modalità d'azione primaria (PMOA) determina se il prodotto è regolamentato dal Center for Drug Evaluation and Research (CDER), dal Center for Biologics Evaluation and Research (CBER) o dal Center for Devices and Radiological Health (CDRH).
-
Percorsi normativi
- Domanda di nuovo farmaco (NDA) o di licenza biologica (BLA): Necessaria se il farmaco o il componente biologico determina l'effetto terapeutico.
- Approvazione pre-market (PMA) o 510(k): Applicabile se il componente del dispositivo è primario (ad esempio, microneedles che facilitano la somministrazione di farmaci).
- I MNP seguono spesso il percorso NDA/BLA a causa della loro funzione farmaco-centrica.
-
Test preclinici e clinici
- Studi preclinici: Includono test di irritazione cutanea, permeazione e stabilità per valutare la sicurezza e l'efficacia della somministrazione.
- Sperimentazioni cliniche: Gli studi di fase I-III valutano la farmacocinetica, l'efficacia e gli effetti avversi nell'uomo.
-
Standard di qualità e produzione
- Conformità con le attuali buone pratiche di fabbricazione (cGMP) per i componenti dei farmaci e dei dispositivi.
- La garanzia di sterilità e i test di stabilità sono fondamentali per i cerotti con i prodotti biologici.
-
Etichettatura e sorveglianza post-vendita
- Le etichette devono includere istruzioni d'uso, avvertenze e condizioni di conservazione.
- I requisiti post-market includono la segnalazione di eventi avversi e potenziali studi di Fase IV.
Per gli acquirenti, la comprensione di questi requisiti garantisce l'allineamento con i fornitori conformi alla FDA, riducendo i rischi di prodotti non conformi. Avete considerato l'impatto di queste normative sulle vostre tempistiche di approvvigionamento o sui criteri di selezione dei fornitori? L'interazione tra sorveglianza dei dispositivi e dei farmaci determina tranquillamente l'affidabilità delle moderne terapie transdermiche.
Tabella riassuntiva:
| Aspetto chiave | Requisiti normativi |
|---|---|
| Classificazione | Regolamentati come prodotti combinati (dispositivo + farmaco/biologico) dalla FDA. |
| Modalità d'azione primaria | Determina la supervisione da parte del CDER (farmaco), del CBER (biologico) o del CDRH (dispositivo). |
| Percorsi di approvazione | NDA/BLA (guidata dal farmaco) o PMA/510(k) (guidata dal dispositivo). Le MNP seguono in genere la NDA/BLA. |
| Requisiti per i test | Studi preclinici (sicurezza, permeazione) e clinici (fasi I-III). |
| Standard di produzione | Conformità alle cGMP per i componenti dei farmaci e dei dispositivi; garanzia di sterilità per i biologici. |
| Obblighi post-commercializzazione | Segnalazione degli eventi avversi, studi di Fase IV e conformità dell'etichettatura. |
Garantite cerotti transdermici conformi alla FDA per il vostro marchio o la vostra rete di distribuzione.
Collaborate con
Enokon
un produttore di fiducia di cerotti transdermici e cerotti per il dolore di alta qualità. La nostra esperienza nella ricerca e sviluppo personalizzata e nella produzione conforme alle normative garantisce una perfetta integrazione nella vostra catena di fornitura.
Contattateci oggi stesso
per discutere i requisiti del vostro progetto e semplificare il processo di approvvigionamento.
Guida Visiva
Prodotti correlati
- Cerotto antidolorifico in gel al mentolo
- Cerotto antidolorifico di medicina al mentolo Icy Hot
- Cerotti riscaldanti antidolorifici per i crampi mestruali
- Cerotto per la tosse e il dolore da asma per adulti e bambini
- Cerotto antidiarroico medicato a base di erbe per il sollievo dell'apparato digerente
Domande frequenti
- Come si comportano i cerotti antidolorifici rispetto ad altri metodi per alleviare il dolore?Scoprite un sollievo mirato e duraturo
- Quando si dovrebbe considerare l'uso di prodotti antidolorifici come creme e cerotti?Ottimizzare il comfort e la sicurezza
- Cosa sono i cerotti antidolorifici e come funzionano?Scoprite la gestione non invasiva del dolore
- Quali sono i diversi tipi di cerotti antidolorifici disponibili?Trovate la soluzione giusta per il vostro dolore
- In che modo i cerotti antidolorifici forniscono un sollievo mirato?Scoprite la scienza alla base di una gestione efficace del dolore